Voici à quoi ressemble la vie avec l'EB
Qu'est-ce que l'EB ?
- Qu'est-ce que l'Epidermolyse Bulleuse ?
- Que signifie ce nom ?
- Une maladie héréditaire
- Comment établit-on un diagnostic ?
- Pourquoi existe-t-il autant de sous-types ?
- Quels sont les symptômes?
- Est-ce douloureux ?
- Comment l'EB est-elle traitée?
- Combien de personnes sont-elles atteintes de l'EB ?
- Littérature
Qu'est-ce que l'Epidermolyse Bulleuse ?
L’épidermolyse bulleuse (EB) est une affection dermatologique génétique rare.
L’atteinte cutanée est présente à la naissance. Une atteinte muqueuse est possible. Il existe 4 grands roupes d’épidermolyses bulleuses: les formes dites simples, les formes dites jonctionnelles, les formes dites dystrophiques et le syndrome de Kindler.
La fragilité cutanée est due à une anomalie d’une protéine de la peau, absente ou non fonctionnelle, elle ne remplit plus son rôle essentiel de lien entre différentes couches de la peau.
Dans le cas de l'EB simple, les bulles se forment en surface, c'est-à-dire au niveau de l'épiderme. Dans le cas de l'EB jonctionnelle, la zone bulleuse se trouve juste en-dessous de l'épiderme et dans le cas de l'EB dystrophique, la formation des bulles se fait au point d'intersection entre l’épiderme et le derme.
La gravité de cette affection est très variable. Elle est déterminée d'une part par la profondeur des bulles et d'autre part par l'étendue des cicatrices. Elle peut varier d'une forme peu sévère à des formes mutilantes, voire mortelles.
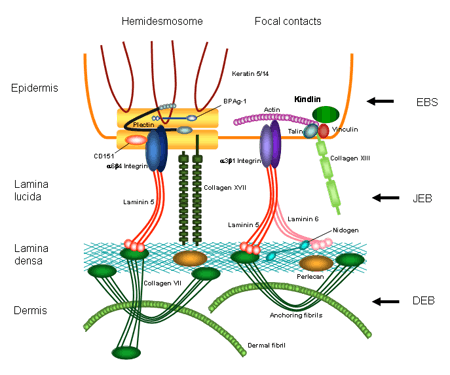
Vue en coupe de la peau humaine
Source: http://www.netzwerk-eb.de/
Toutes les formes d'EB sont héréditaires mais le gène responsable de cet accident génétique a été identifié pour la plupart. Cet accident génétique provoque une destruction des cellules dermiques ou une adhésion insuffisante des différentes couches de l'épiderme. La peau atteinte est extrêmement fragile et se décolle au moindre frottement, créant ainsi en permanence une formation de bulles, caractéristique typique de l'Epidermolyse Bulleuse.
Que signifie ce nom ?
- La peau se compose de différentes couches: la couche externe ou l’épiderme et la couche interne ou le derme.
- 'Bulleuse' est le nom scientifique donné aux bulles.
- 'Lysis' signifie détruire ou décoller
L’Epidermolyse Bulleuse est donc la destruction de l'épiderme caractérisée par une formation de bulles.
Une maladie héréditaire
L'EB n'est pas contagieuse, elle est héréditaire. Les parents transmettent la maladie à l’enfant.
La plupart des formes d’EB simples sont transmises sur le mode dominant, les formes dystrophiques sur le mode récessif.
Une maladie à transmission dominante est due à une anomalie d’un gène d’une paire de gènes. Un des parents est également atteint de la maladie. Le risque que l’enfant transmette la maladie à la génération suivante est de 50%.
Dans certains cas aucun des parents n’est atteint mais le défaut se manifeste pour la première fois chez l’enfant. Il s’agit alors d’une néo-mutation génétique (ou mutation de novo).
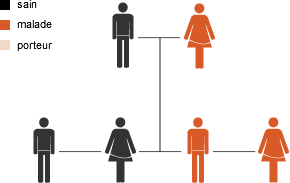
Hérédité dominante
Une maladie à transmission récessive est due à une anomalie des deux gènes d’une paire de gènes et ne peut être transmise que si les deux parents de l’enfant portent le gène défectueux (sans le savoir). Si les deux parents sont porteurs, le risque que l’enfant soit atteint est de 25%.
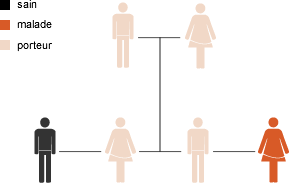
Hérédité récessive
Les familles qui se posent des questions sur l'hérédité peuvent demander une consultation génétique (remboursée par la mutuelle) dans un des 8 Centres de Génétique Humaine, liés aux hôpitaux universitaires.
Comment établit-on un diagnostic ?
Au moyen d’une biopsie
Il est essentiel de réaliser des prélèvements dans les plus brefs délais. Il faut établir un diagnostic correct et déterminer le sous-type de l’EB afin de pouvoir fournir des informations précises aux parents. Les biopsies sont utilisées pour le mapping-immunofluorescence (coloration avec les anticorps) et la microscopie électronique.
Il est crucial que ces prélèvements soient effectués correctement et qu’il soit établi au préalable vers quel laboratoire, de quelle manière et à quel moment ils devront être transportés. En cas de doute, il convient de faire intervenir un centre spécialisé.
Au moyen d’une analyse ADN
Dès qu’on s’est fait une idée du type et du sous-type d’EB, une analyse moléculaire du gène muté pourra être réalisée.
Une fois que la mutation aura été identifiée, il sera possible de vérifier pour les formes récessives si les parents et les autres membres de la famille sont porteurs du gène. Par ailleurs, lors d’un prochain projet parental, il pourra être procedé à un diagnostic préimplantatoire ou à un diagnostic génétique prénatal.
En lire plus :
Le mendéliome et le séquençage à haut débit
Laboratory Diagnosis (Debra International, en anglais uniquement)
Pourquoi existe-t-il autant de sous-types ?
La maladie peut prendre des formes très diverses, mais, de manière globale, on peut distinguer quatre types majeurs :
- L’EB simple (EBS, clivage dans la couche basale)
- L’EB jonctionnelle (EBJ, clivage dans la lamina lucida de la membrane basale)
- L’EB dystrophique (EBD, clivage en-dessous de la lamina densa)
- Le syndrome de Kindler (clivage à un niveau variable)
De nombreux sous-types peuvent encore être distingués au sein de ces quatre types majeurs. La gravité du sous-type est conditionnée par le niveau de clivage (plus le clivage est profond, plus le niveau de gravité est élevé), par le type de mutation et en conséquence par l’absence (type grave) de la protéine exprimée ou par le niveau de fonctionnalité réduite (type moins grave) de celle-ci.
Plus d’informations au sujet de la classification
Quels sont les symptômes?
Chaque type d’EB a ses propres variantes symptomatiques.
Les formes légères se manifestent surtout par des bulles aux mains et/ou aux pieds, mais parfois aussi sur tout le corps, ainsi que dans la bouche et le pharynx. Ces bulles qui guérissent sans laisser de cicatrice, peuvent être très douloureuses. Le quotidien est marqué par de nombreuses gênes telles que le besoin de soins journaliers, frottement des vêtements, chaussures non adaptées, mobilité limitée etc…
Les formes plus sévères se manifestent généralement par des bulles sur le corps entier. Les bulles laissent des cicatrices et peuvent provoquer la fusion des doigts et orteils. Le risque d’infection est omniprésent du fait de la chronicité des plaies. Ces formes peuvent également atteindre les muqueuses de la bouche et de l’oesophage, rendant l’alimentation diffi cile et ralentissant la croissance. L’oeil
peut aussi être affecté. A l’âge adulte, les plaies chroniques peuvent évoluer vers des cancers très agresssifs de la peau. Toutes ces complications peuvent entraîner une grande invalidité et une espérance de vie limitée.
Est-ce douloureux ?
La douleur existe en permanence, mais elle varie en fonction du type d’EB. Chacun d’entre nous connaît la douleur causée par une simple ampoule. On peut dès lors imaginer les souffrances quotidiennes d’une personne atteinte d’épidermolyse bulleuse dont les bulles sur tout le corps peuvent apparaître par le simple frottement des vêtements, un pansement déplacé, ou même sans raison.
Les patients EB souffrent souvent aussi de prurit chronique. Le patient a tendance à se gratter, ce qui ne fait que provoquer de nouvelles bulles et/ou plaies.
Comment l'EB est-elle traitée?
A l’heure actuelle, il n’y a aucune possibilité de traitement.
Les soins infirmiers sont la pierre d’angle de la prise en charge médicale. Afin d’éviter toutes infections ou complications, les soins journaliers consistent à percer les nouvelles bulles avec des aiguilles stériles. Ensuite les plaies sont désinfectées et recouvertes de crèmes, compresses et pansements. Ces soins peuvent durer des heures.

Les pansements adhérents sont à proscrire !
Les soins peuvent durer de longues heures pour les patients atteints d'une forme grave d'EB et sont très lourds tant pour eux que pour leur famille. En outre, ces soins représentent une charge financière importante. Chaque cas étant différent, c'est le patient qui sait lui-même comment se soigner au mieux. L'état de santé du patient évolue en fonction du temps, du stress ou d’autres facteurs.
Pour davantage d’informations, vous pouvez consulter la rubrique 'Soins'.
Une consultation pluridisciplinaire incluant un soutien psychologique de l’enfant et de sa famille est nécessaire.
Les récentes découvertes scientifiques ont contribué à une meilleure compréhension de l’anomalie génétique et offrent des perspectives :
• les gènes responsables de la plupart des formes d’EB sont maintenant connus
• un diagnostic prénatal précoce est possible, y-compris le diagnostic pre-implantatoire (FIV)
• dans un avenir plus lointain : la thérapie protéïque, la thérapie cellulaire et la thérapie génique (en lire plus dans la rubrique "recherche scientifique")
Combien de personnes sont-elles atteintes de l'EB ?
L’EB est une maladie rare et les formes les plus sévères le sont encore davantage. En Belgique, on estime la fréquence de l’EB à 3 0 4 naissances par an (1 sur 22.000).
Il y aurait environ 500.000 cas dans le monde, hommes et femmes confondus.
Littérature
Enfants papillons, informations destinées aux patients, UZ Leuven
L'épidermolyse bulleuse (Dermatologie Actualité 2012, n°130)
M. Morren, M. Flour, M. Garmijn - Département de dermatologie, UZ Leuven
Encyclopédie Orphanet Grand Public Les épidermolyse bulleuse héréditaires
Inherited epidermolysis bullosa (en anglais - Orphanet Journal of Rare Diseases 2010, 5:12)
Jo-David Fine - Departments of Medicine (Dermatology) and Pediatrics Vanderbilt University School of Medicine, and Head, National Epidermolysis Bullosa Registry Nashville, TN, USA
Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification (en anglais - published online March 31, 2014 - Journal of American Academy of Dermatology), Jo-David Fine et al.
