Zo voelt het leven als je EB hebt
Wat is EB ?
- Wat is epidermolysis bullosa ?
- Wat betekent de naam ?
- Hoe krijg je EB ?
- Hoe wordt de diagnose van EB gesteld?
- Waarom zovele subtypes?
- Wat zijn de symptomen van EB ?
- Is EB pijnlijk ?
- Hoe wordt EB behandeld ?
- Hoeveel mensen hebben EB?
- Bijkomende literatuur
Wat is epidermolysis bullosa ?
Epidermolysis bullosa (EB) is de verzamelnaam voor een groep van zeldzame, erfelijke blaaraandoeningen waarbij er blaren ontstaan bij de minste wrijving of aanraking, op de huid maar soms ook op de slijmvliezen.
De oorzaak is een aangeboren fout in de huideiwitten, waardoor de verschillende huidlagen niet goed aan elkaar vastzitten. Ze komen gemakkelijk los en zo ontstaan er blaren en open wonden.
| EB is zeldzaam | EB is complex | EB is een erfelijke ziekte | EB is levensbeperkend |
| 500.000 mensen hebben EB wereldwijd |
4 hoofdtypen: EB Simplex (EBS) Dystrofische EB (DEB) Junctionele EB (JEB) Kindler EB (KEB) |
20 betrokken genen zijn gekend en duizenden mutaties | EB heeft fysieke en emotionele impact op het dagelijkse leven |
| Prevalentie: 1 op 30.000 mensen hebben EB | 30 verschillende klinische subtypen | Bijkomend ook ’modifier’- genen die de ernst en symptomen beïnvloeden | Typische kenmerk is extreme fragiliteit van de huid en slijmvliezen |
| EB komt voor bij mannen en vrouwen, in alle etnische groepen | Grote verschillen in symptomen en ernst tussen de EB-typen |
EB kan dominant of recessief overgeërfd worden, afhankelijk van de mutatie. |
Steeds terugkerende blaarvorming leidt in de meeste gevallen tot chronische wonden, fibrose en pijn. |
|
Dominant: bijna alle EBS en DDEB (dominante DEB) |
Bij sommige zeer ernstige subtypen leiden infectie, groeifalen en orgaanfalen tot vroegtijdig overlijden. | ||
|
Recessief: de meeste JEB, RDEB (recessieve EB) en Kindler syndroom |
Mensen met de meer ernstige vormen ontwikkelen een agressieve vorm van huidkanker. |
Wat betekent de naam ?
- De huid bestaat uit verschillende lagen; de bovenste laag noemen we de opperhuid of 'epidermis' (eronder ligt de 'dermis').
- 'Bullosa' is de wetenschappelijke naam voor een 'blaar'.
- 'Lysis' betekent het 'loslaten'.
Epidermolysis Bullosa betekent dus het loslaten van de opperhuid met blaarvorming als gevolg.
Hoe krijg je EB ?
EB is niet besmettelijk, maar erfelijk. Ouders geven de genetische fout door aan hun kinderen.
De milde vormen worden meestal dominant doorgegeven van ouder op kind (één van de ouders heeft EB) en de meer ernstige vormen worden meestal recessief doorgegeven (beide ouders zijn drager van het afwijkende gen maar zijn gezond).
Families met vragen over erfelijkheid kunnen terecht voor genetische raadpleging in het centrum voor menselijke erfelijkheid, verbonden aan het EB-centrum in UZ Leuven.
Hoe wordt de diagnose van EB gesteld?
huidbiopten
Huidbiopten zijn nodig voor immunofluorescentie-mapping (kleuring van antistoffen) en elektronenmicroscopie om het juiste subtype te kunnen bepalen.
Het is cruciaal dat biopten op een correcte manier worden afgenomen en dat vooraf bepaald wordt naar welk labo en opwelke manier en tijdstipze dienen getransporteerd te worden. Bij twijfel laat men dit beter over aan een centrum dat de ervaring heeft om dit te doen.
DNA-onderzoek
Eens men een idee heeft over het type en subtype van EB is moleculair onderzoek naar het gemuteerde gen mogelijk.
Wanneer de mutatie gekend is, kan bij recessieve vormen dragerschap bij ouders en familieleden worden opgezocht en bestaat de mogelijkheid voor pre-implantatie of prenatale genetische diagnostiek bij een volgende kinderwens.
Meer lezen:
Mendelioomanalyse in UZ Leuven
Laboratory Diagnosis (Debra International, voorlopig enkel in het Engels)
Waarom zovele subtypes?
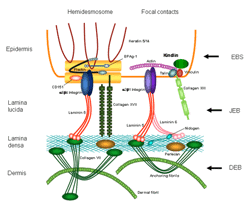
http://www.netzwerk-eb.de/
De aandoening kan zeer uiteenlopende vormen aannemen, maar globaal onderscheidt men vier hoofdgroepen:
- EB simplex (EBS, splitsing in de basale laag)
- junctionele EB (JEB, splitsing in de lamina lucida van de basale membraan)
- dystrofische EB (DEB, splitsing onder de lamina densa)
- Kindlersyndroom (splitsing op variabel niveau)
Binnen deze hoofdgroepen zijn nog talrijke subtypen te onderscheiden. De ernst van het subtype hangt af van het splitsingsniveau (hoe dieper, hoe ernstiger) en het type van mutatie en bijgevolg het afwezig zijn (ernstige vorm) of de mate van minder functioneel zijn (minder ernstige vorm) van het geëxprimeerde eiwit.
Wat zijn de symptomen van EB ?

foto: UZ leuven, neonatologie
Blaarvorming is het typische kenmerk van EB. De aandoening kan echter zeer uiteenlopende vormen aannemen naargelang het subtype.
Bij mildere vormen beperkt de blaarvorming zich tot de handen en voeten. De blaren kunnen zeer talrijk en pijnlijk zijn, maar ze genezen zonder littekens.
Bij meer ernstige vormen treden de blaren op over het ganse lichaam en vaak ook inwendig op de slijmvliezen (mond- en keelholte, spijsverteringsstelsel, ogen). De huid is zeer gevoelig en de minste beweging (zich omdraaien in bed) of aanraking (kleding) kan blaren en open wonden veroorzaken. De blaren genezen moeilijk en veroorzaken littekens en vergroeiingen van de vingers en tenen. Mond- en slokdarmletsels veroorzaken ernstige voedings- en groeiproblemen.
De meeste mensen met EB hebben constant last van pijn of jeuk. Sommige patiënten ontwikkelen huidkanker en hun levensverwachting is dan ook laag.
Is EB pijnlijk ?
Pijn is bij EB constant aanwezig, maar varieert naargelang het subtype EB. Iedereen heeft wel eens last van pijn die veroorzaakt wordt door één blaar, bv. na het dragen van nieuwe schoenen. Stel je voor dat je elke dag weer nieuwe blaren ontdekt, over het ganse lichaam, gewoon door de druk van kleding, of een verschoven verband, of zomaar...
De meeste mensen met EB hebben ook constant last van jeuk op plaatsen van genezende wonden. Door het krabben ontstaan weer nieuwe blaren en open wonden.
Hoe wordt EB behandeld ?
Geen genezing, wel behandeling.
EB kan voorlopig niet genezen worden. Het hoofddoel van de behandeling is het ontstaan van blaren zoveel mogelijk te voorkomen en bestaande blaren en letsels zo snel mogelijk te doen genezen.
Verder is het ook belangrijk om infecties te voorkomen of te genezen en de algemene gezondheid te ondersteunen door o.a. een aangepast dieet.
Elke patiënt heeft een persoonlijk verzorgingsplan dat wordt aangepast in functie van de exacte diagnose, de locatie van de wonden, en ook de persoonlijke voorkeur van de patient en zijn verzorger. Oudere kinderen en volwassenen weten perfect hoe ze de wondzorg wensen (frequentie van de verbandwissel, al dan niet dagelijks een volledige verbandwissel, al dan niet een dagelijks bad, enz.) en zijn experts inzake technieken en soorten verbanden.
Een multidisciplinaire aanpak is aangewezen, alsook psychosociale begeleiding van het kind en zijn familie.
Voor meer gedetailleerde informatie kan u terecht in de rubriek 'Verzorging'.
Een EB-huid verdraagt geen gewone huidpleisters!
Toekomstmuziek?
De recente ontwikkelingen in het wetenschappelijk onderzoek hebben geleid tot een beter inzicht in het ontstaan van de aandoening en bieden perspectieven:
• voor de meeste vormen van EB weet men in welk gen de fout zit;
• prenatale diagnose is mogelijk, ook pre-implantatie genetische diagnose (PGD na IVF-behandeling);
• nieuwe behandelingstechnieken in de toekomst: proteïnetherapie, celtherapie of gentherapie (meer hierover in de rubriek wetenschappelijk onderzoek).
Hoeveel mensen hebben EB?
EB is een zeldzame ziekte en de ernstige vormen zijn nog zeldzamer. Men schat dat EB zich voordoet in een verhouding van 1 op 22.000. In België zou het gaan om 4 à 5 geboortes per jaar. Wereldwijd treft EB ongeveer 500.000 mensen, zowel mannen als vrouwen.
Bijkomende literatuur
Infografiek (Debra International) Wat is EB?
Folder informatie voor patiënten van UZ Leuven
Hereditaire epidermolysis bullosa (Dermatologie Actueel 2012, nr. 130) M. Morren, M. Flour, M. Garmijn - Afdeling Huidziekten, UZ Leuven
Epidermolysis Bullosa (informatie op de website van UMCG Expertcentrum voor blaarziekten, Groningen)
EB-Handboek (mei 2020): In overleg met het Centrum voor Blaarziekten (UMCGroningen) heeft DEBRA Nederland een aantal hoofdstukken uit het EB Handboek van het EB Centrum uit Oostenrijk (EB-Haus in Salzburg) laten vertalen in het Nederlands: Eerste hulp - Wondverzorging - Huidkanker - Dagelijks leven - Beweging en Sport - Voeding.
